The plot_tox_endpoints function creates a set of boxplots representing EAR
values for each endPoint based on the selected data. A subset of data is first
chosen by specifying a group in the filterBy argument. The
filterBy argument must match one of the unique options in the category.
For example, if the category is "Chemical Class", then the filterBy argument
must be one of the defined "Chemical Class" options such as "Herbicide".
A boxplot is generated for each endPoint. The EAR values that are used to
create the boxplots are the mean or maximum (as defined by mean_logic) for each
site as described in "Summarizing the data"in the Introduction vignette.
Usage
plot_tox_endpoints(
chemical_summary,
category = "Biological",
filterBy = "All",
manual_remove = NULL,
hit_threshold = NA,
mean_logic = FALSE,
sum_logic = TRUE,
font_size = NA,
title = NA,
x_label = NA,
palette = NA,
top_num = NA
)Arguments
- chemical_summary
Data frame from
get_chemical_summary.- category
Either "Biological", "Chemical Class", or "Chemical".
- filterBy
Character. Either "All" or one of the filtered categories.
- manual_remove
Vector of categories to remove.
- hit_threshold
Numeric threshold defining a "hit".
- mean_logic
Logical.
TRUEdisplays the mean sample from each site,FALSEdisplays the maximum sample from each site.- sum_logic
logical.
TRUEsums the EARs in a specified grouping,FALSEdoes not.FALSEmay be better for traditional benchmarks as opposed to ToxCast benchmarks.- font_size
Numeric to adjust the axis font size.
- title
Character title for plot.
- x_label
Character for x label. Default is NA which produces an automatic label.
- palette
Vector of color palette for fill. Can be a named vector to specify specific color for specific categories.
- top_num
Integer number of endpoints to include in the graph. If NA, all endpoints will be included.
Details
Box plots are standard Tukey representations. See "Box plot details" in the Basic Workflow vignette. for more information.
Examples
# This is the example workflow:
path_to_tox <- system.file("extdata", package = "toxEval")
file_name <- "OWC_data_fromSup.xlsx"
full_path <- file.path(path_to_tox, file_name)
tox_list <- create_toxEval(full_path)
ACC <- get_ACC(tox_list$chem_info$CAS)
ACC <- remove_flags(ACC)
cleaned_ep <- clean_endPoint_info(end_point_info)
filtered_ep <- filter_groups(cleaned_ep)
chemical_summary <- get_chemical_summary(tox_list, ACC, filtered_ep)
# \donttest{
plot_tox_endpoints(chemical_summary,
filterBy = "Cell Cycle",
top_num = 10
)
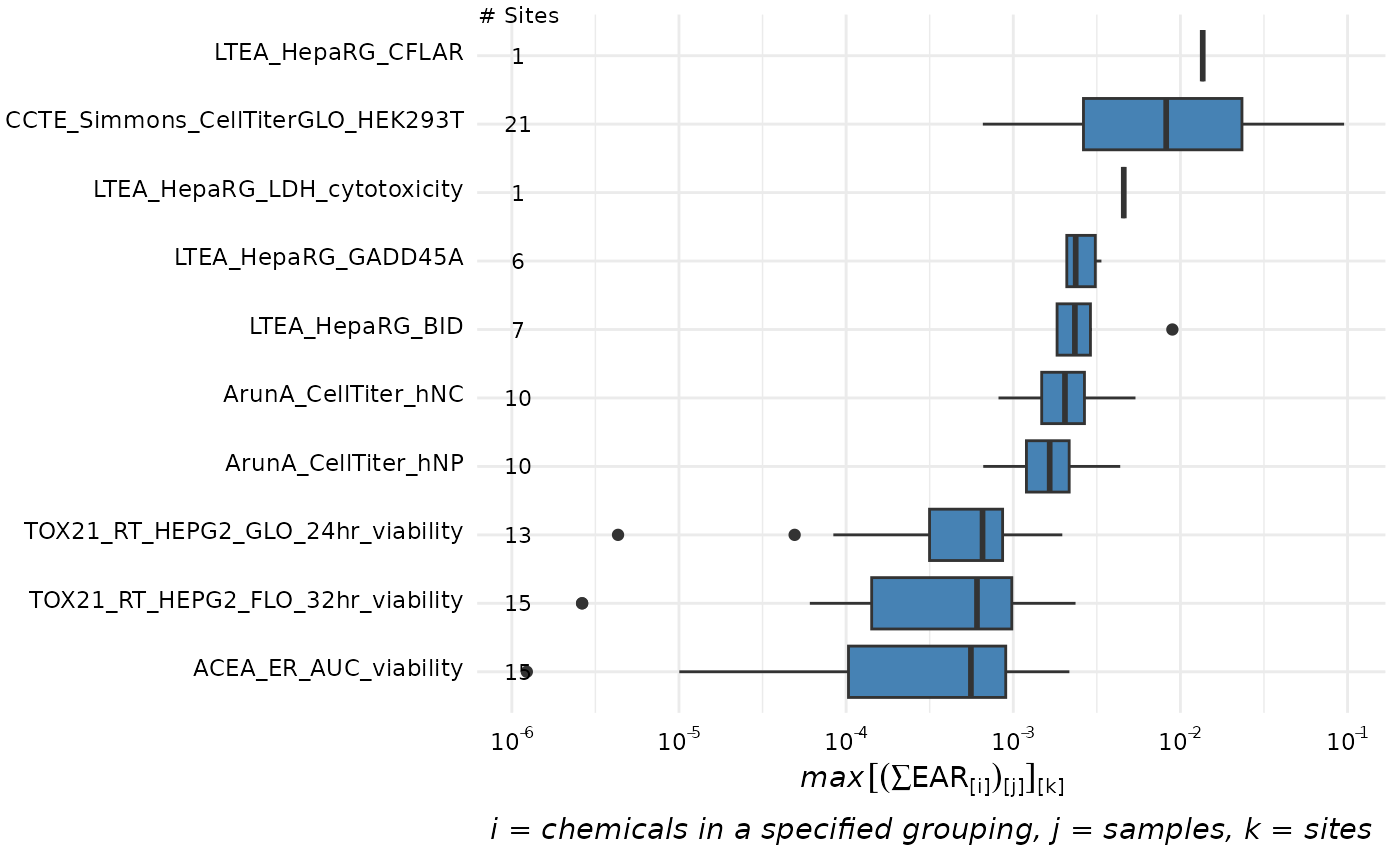 plot_tox_endpoints(chemical_summary,
filterBy = "Cell Cycle",
top_num = 10,
x_label = "EAR"
)
plot_tox_endpoints(chemical_summary,
filterBy = "Cell Cycle",
top_num = 10,
x_label = "EAR"
)
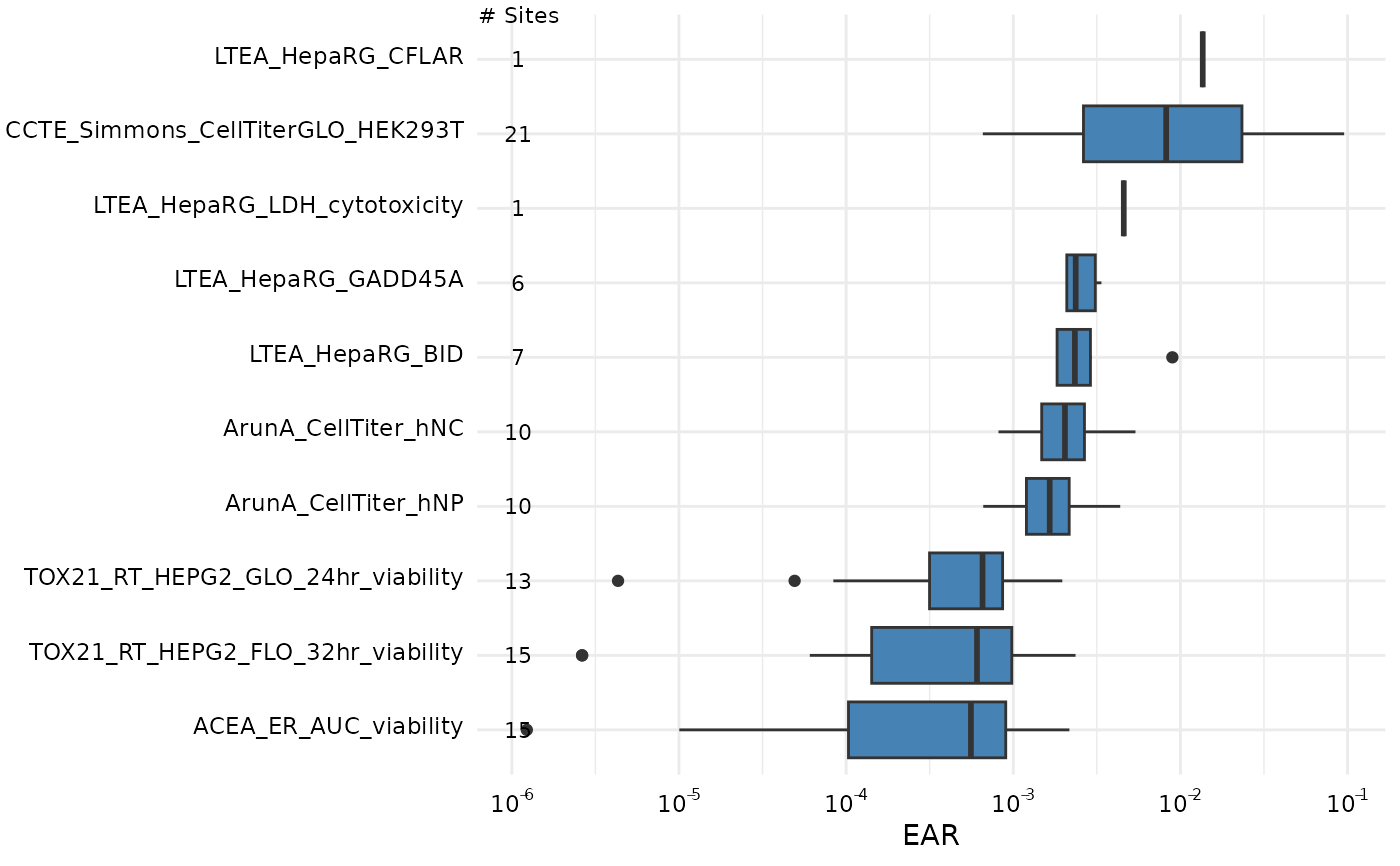 plot_tox_endpoints(chemical_summary,
category = "Chemical Class", filterBy = "PAHs",
top_num = 10, hit_threshold = 0.001
)
plot_tox_endpoints(chemical_summary,
category = "Chemical Class", filterBy = "PAHs",
top_num = 10, hit_threshold = 0.001
)
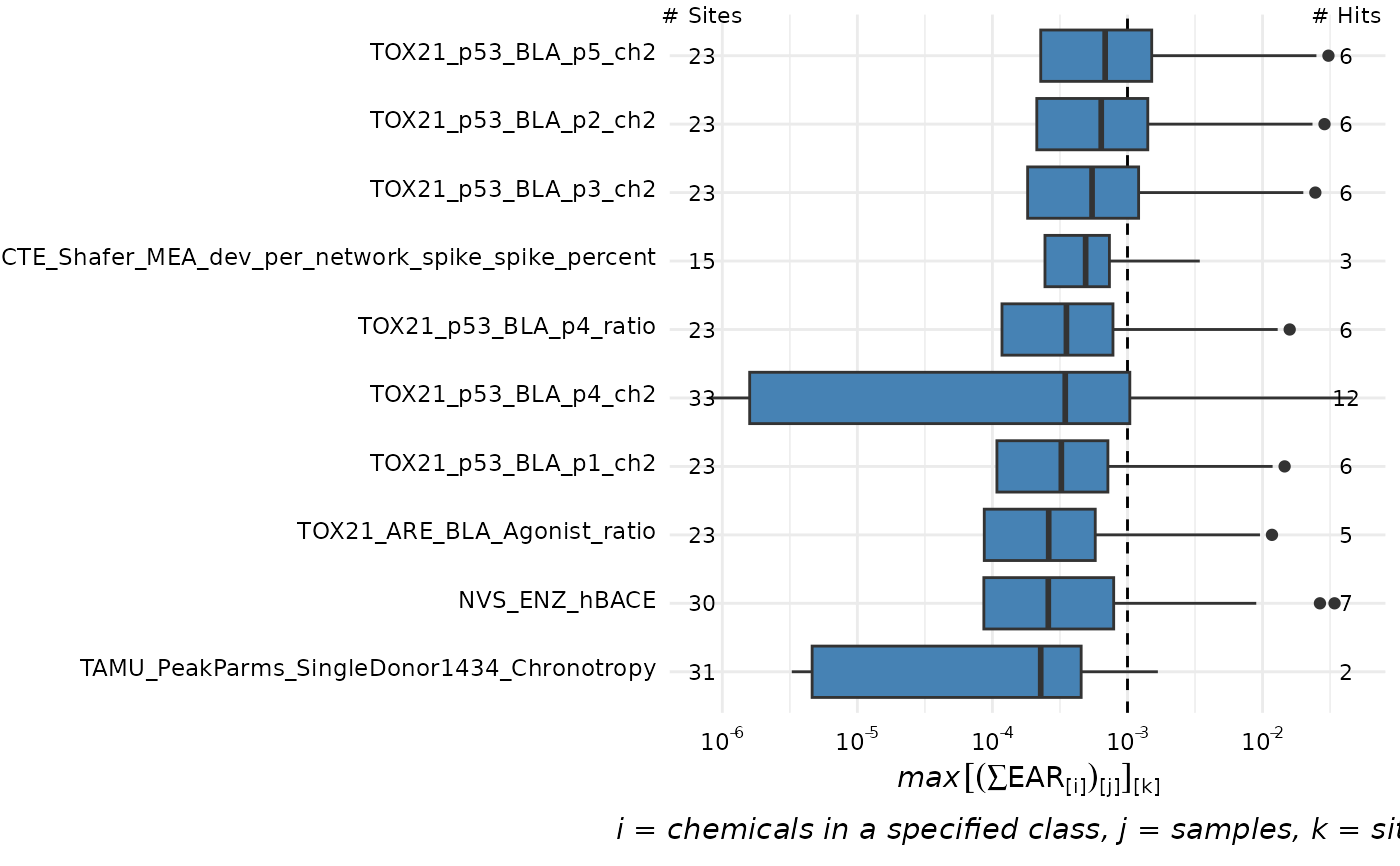 plot_tox_endpoints(chemical_summary, category = "Chemical", filterBy = "Atrazine")
plot_tox_endpoints(chemical_summary, category = "Chemical", filterBy = "Atrazine")
 plot_tox_endpoints(chemical_summary, category = "Chemical", top_num = 10)
plot_tox_endpoints(chemical_summary, category = "Chemical", top_num = 10)
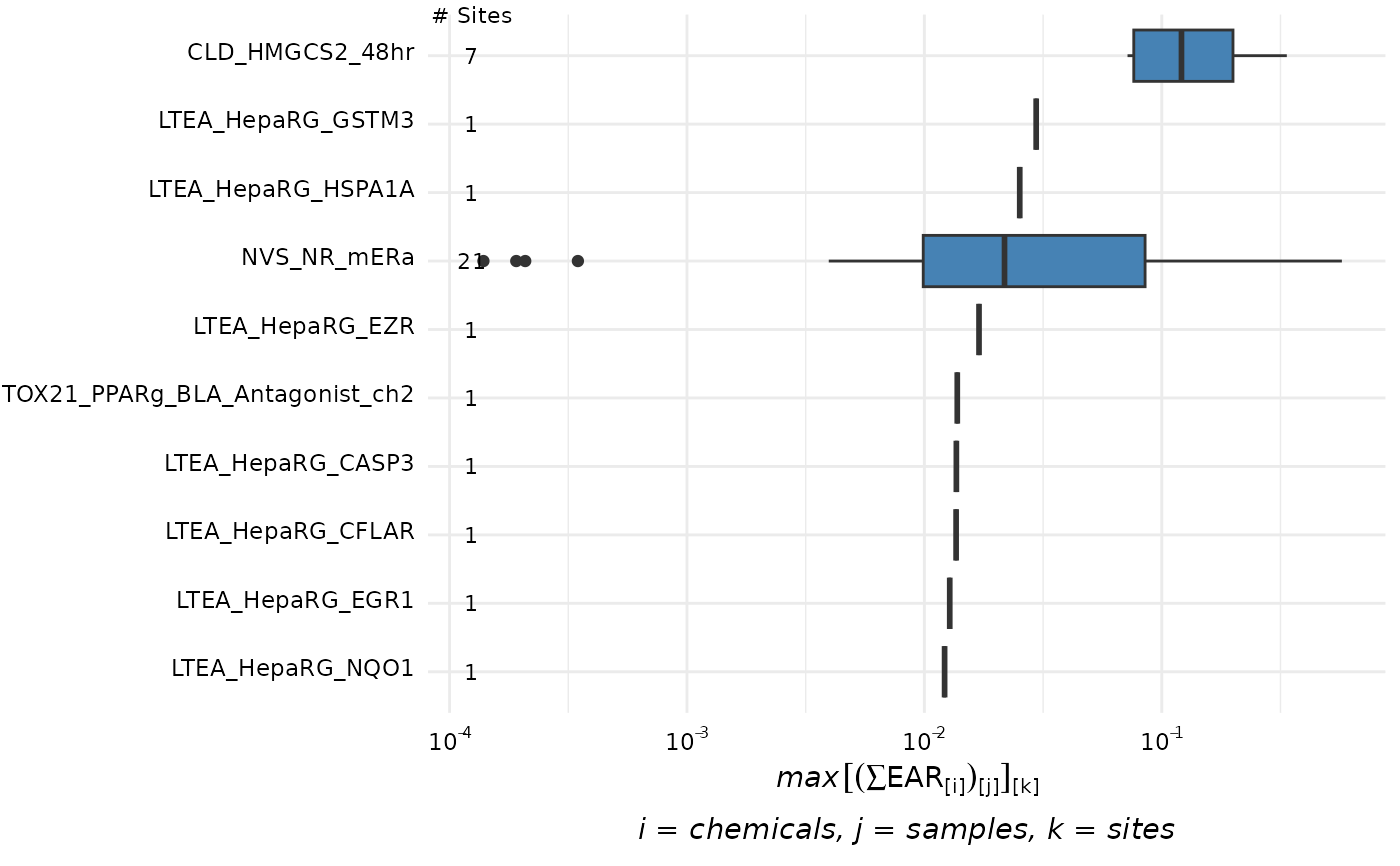 single_site <- dplyr::filter(chemical_summary, site == "USGS-04024000")
plot_tox_endpoints(single_site, category = "Chemical", top_num = 10)
single_site <- dplyr::filter(chemical_summary, site == "USGS-04024000")
plot_tox_endpoints(single_site, category = "Chemical", top_num = 10)
 # }
# }